
Journal Club

2020年 6月8日(担当:若松)
Science 368, eaat3987 (2020), DOI: 10.1126/science.aat3987
Interleukine-13 drives metabolic conditioning of muscle to endurance exercise
筋肉の収縮時に筋肉組織から産生されるサイトカイン、およびペプチドはMyokineと呼ばれ、autocrineによって筋肉の代謝の制御に寄与している。また、筋肉だけでなく、para/endocrineによって脂肪組織、肝臓、脳などの他の組織にも影響を及ぼす。IL-6はmyokineの中で最も研究されており、筋収縮に応じて産生され、グルコースの取り込みや脂肪酸の酸化を促進する。しかしながら、持続的に運動をしている人では、定常状態での血漿中IL-6の濃度は低く、また肥満の人で増加する。これらのことから、持続的な運動によって誘導される筋肉の変化はIL-6以外のmyokineによって制御される可能性が考えられた。著者らは、肥満患者、健常人、運動している人の血漿中のTh1/Th2サイトカインを網羅的に解析することで、運動している人で血漿中のIL-13の濃度が高いことを見出した。持続的な運動は筋肉組織のILC2を増加させ、ILC2からのIL-13産生を増加させていた。また、ILC2の増加およびIL-13産生の増強は、持続的な運動によって血漿中のIL-33濃度の増加が起因している可能性を示唆している。RNAseq解析から、IL-13が脂肪酸のβ-oxidation関連遺伝子、およびグリコーゲン貯蔵関連遺伝子の発現制御に寄与していることが明らかとなった。RNAseq解析の結果と一致して、IL-13は筋肉組織への脂肪酸の取り込み、中性脂肪の消費、およびグリコーゲンの貯蔵を促進していた。持続的な運動はミトコンドリアの生成を促進することが知られている。そのため、著者らはミトコンドリア関連遺伝子の発現をRNAseqデータを用いて解析した。その結果、電子伝達系を含めたほとんどのミトコンドリ関連遺伝子がIL-13欠損マウスで低下していた。また、運動によるミトコンドリア生成もIL-13欠損マウスで異常が起こっていた。これらの結果と一致して、持続的な運動によって起こる電子伝達経のC-IIおよびC-IVの活性化がIL13欠損マウスで認められなかった。これらのIL-13を介した筋肉の代謝制御はIL-13下流のcanonicalな転写因子であるSTAT6ではなく、STAT3を介して起こっていることを明らかにしている。これらのことから持続的な運動によって産生されたIL-13はSTAT6を介して筋肉の代謝リプログラミングを制御することで、筋肉の質的変化を誘導することが示唆される。
免疫学的にこの論文を見たときに、運動を続けている人はアレルギーや喘息になりやすいのではないかと考えるのではないだろうか。特にこの点に関しては言及していないが、病変を発症させるためのIL-13を含めたTh2サイトカインの濃度と比較すると少ないのかもしれない。また、持続的な運動は抗原特異的な免疫応答を誘導しないために、上記した疾患には繋がらないのかもしれない。一方で、Treg除去や抗PD1/CTLA4抗体投与などで免疫チェックポイント機構を失わせると疾患の発症が起こる可能性はあるだろう。
2020年 6月1日(担当:町山)
Nat Biotechnol., 2020 Mar 23. doi:
10.1038/s41587-020-0462-y.
Human Chimeric Antigen Receptor Macrophages for Cancer
Immunotherapy.
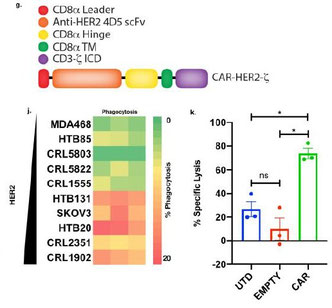
キメラ抗原受容体CARをマクロファージに遺伝子導入したCAR-Mが固形がんに対する抗腫瘍効果を示した論文。CAR-T細胞療法は白血病やリンパ腫などの血液腫瘍には有効だが、固形腫瘍には目立った成果が報告されていない。その原因として「がん微小環境」において腫瘍随伴マクロファージTAMが免疫抑制を誘導し腫瘍の成長を手助けしていることが挙げられる。著者らは、HER2をターゲットにしたCARをAd35アデノウイルス感染によってヒトマクロファージに導入した“CAR-M”を作成した。CAR-MはHER2発現依存的な腫瘍細胞の貪食作用があり、炎症型のM1タイプに似た遺伝子プロファイルとタンパク質発現を示していた。NSGヒト化マウスにCAR-Mを移入することで、腫瘍の排除と生存率の延長を確認している。TAMは主に抗炎症型のM2マクロファージであって、新たにがん組織に浸潤してくるマクロファージをM2にしてがん微小環境をさらに助長するが、CAR-Mはがん組織に侵入してもM1タイプ様を維持しており、さらにがん組織内のマクロファージもM2からM1様の発現パターンに変えてしまうようだ。CAR-Mはがん抗原特異的なT細胞を腫瘍組織にリクルートして獲得免疫系の活性化も促し、腫瘍組織内の抗腫瘍効果を高める環境を作り出す。問題点としては、CAR-Mが腫瘍組織以外にもリクルートされることがあり、そういった正常組織内においてもM1様の性質が維持されることで、周囲のマクロファージをM1タイプへ転換させ炎症を惹起させることが挙げられる。M2に分化しないってことがKeyですが、PC1/PC2プロファイルを見ると、M1そのものでもなくて、新たに人工的に作り出されるM1に近い細胞群ってことで、完璧ではないようだが、Ad35アデノウイルス感染って方法が、TollやRIG-Iなどを介したinnate signalを惹起させて細胞内微生物に対する免疫応答をベースであげているのかなあ、と思う。
2019年1月15日(担当:矢那瀬)
Science. 2018 Dec 21; 362(6421): 1416-1422.
doi: 10.1126/science.aas9090.
NK cell-mediated cytotoxicity contributes to
tumor control by a cytostatic drug combination.
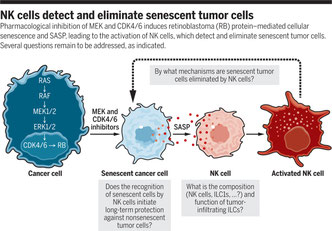
K-Rasの変異による発がんはよくあることで、MEK阻害剤とCDK4/6阻害剤の上下からの挟み撃ちの化学併用療法が有用である。ただ、Lowe研ではこの抗腫瘍効果がNKがないマウス(Anti-NK1.1の投与)では弱いことに気がついた。KrasG12D Trp53-/-の肺がんモデルを用いて、両阻害剤処理・未処理群での遺伝子発現クラスター解析を行ってみると、p53ではなくRbを介する細胞老化Senescenceが起こっており、SASP因子の中でもNKの活性化に寄与する分子が阻害剤処理腫瘍細胞から分泌されていることが分かった。IL-15やIL-6などの炎症性サイトカイン、CCL2やCXCL1などのケモカインも上昇しているが、SASP因子の中でもNF-kBシグナル下流で分泌されるTNFaがNK活性化の鍵となるようだ。Stellate細胞の肝癌発症モデルを始め、これまでSASPは腫瘍増殖に促進的に働く現象と考えれる事例が多かったが、こういった老化に伴う炎症を担う因子は、同時に免疫系の賦活化にも寄与していると考えるべきだろう。ただ、抗がん剤を作用させた腫瘍細胞がSASPとなる現象は、ただのCell cycle arrestとどう異なるのか、SASP自体の提議がいまいち過大解釈される傾向にあることと、SASP因子の中でもTNFaだけしか責任分子でないのだとしたらそれはTNFaと記述すべきかも知れないし、Too muchな印象もある。一方、臨床からの側面では、抗がん剤と免疫賦活療法はDAMPsとSASPを介して自然免疫と、Tumor antigenを介して獲得免疫の両方を活性化できる訳だから、治療の方向性としては益々併用療法の重要性は増していくだろう。また、放射線治療ではSASPは誘導されないのか、なんて考えてしまう。
2018年6月18日(担当:矢那瀬)
Nature. 2018 May;557(7707):724-728.
doi: 10.1038/s41586-018-0119-x.
Microglial control of astrocytes in response to microbial
metabolites.
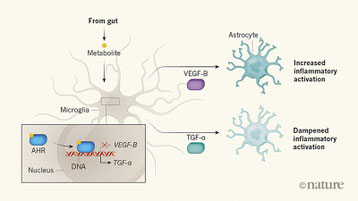
腸内細菌の代謝産物をマイクログリアを感知し、アストロサイトを活性化して脳内炎症を増強する、という話題性のあるAxisで免疫応答を説明している論文。食物中のトリプトファンがアリルハイドロカーボン受容体AHRに結合することは知られていることで、CX3CR1-Creを用いてResidentialなアストロサイトだけAHRを欠損させ、EAEモデルを組んでみた。すると、KOではクリニカルスコアが改善しない。WTとKOのマイクログリアの発現遺伝子解析を行ってみると、どうやらAHRからのシグナルは炎症に対して抑制的に働いているらしい。で、さまざまな制御分子に関してネットワーク解析してみると、AHR活性化によって、TGF-α発現増強、VEGF-B発現低下、がKeyとして働いているようで、VEGF-Bによって炎症性サイトカインなどが分泌され、EAEのスコアリングが上がってしまう。一方、TGF-αは抑制的に働く。トリプトファン欠損食ではそれが起こらず、その代謝産物がオリゴデンドログリアのAHRを介してEAEを抑制する。ヒトのアストロサイトでもトリプトファン代謝産物で炎症性サイトカイン分泌が下がり、多発性硬化症の病巣でも、TGF-αは炎症性サイトカイン分泌に抑制的に、VEGF-Bは促進に働いている。トリプトファン代謝産物がAHRにダイレクトに結合しているのか、論文では代謝産物添加でAHRの発現自体が上昇しているが、AHRの発現上昇自体が重要なのか、AHRと結合するだけでよいのか、またAHRが転写因子としてTGF-αやVEGF-Bのプロモーター領域に結合して転写を調節しているのか、など詳細はこの論文では示されていないが、まあ、トリプトファンを食べて入れば脳炎は抑えられていると言うことなんだろう。
2018年6月4日(担当:町山)
Immunity 2018 May 22. doi:
10.1016/j.immuni.2018.04.018.
Transcription Factor IRF8 Orchestrates
the Adaptive Natural Killer Cell Response
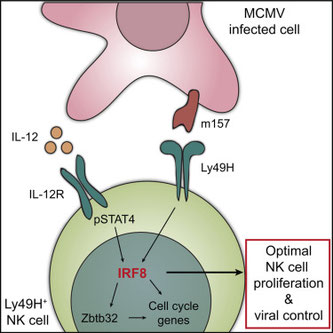
NK細胞とIRFファミリー分子に関しては、これまでもIRF1KOでNK細胞の分化がおかしくなったり、IRF2がサバイバルに必要だったり、知られていたらしい。最近、IRF8の遺伝子変異家系が見つかり、どうやら成熟NK細胞が減少したり細胞傷害活性が低下するとのことで、そのメカニズムを知ろうとした。マウスのMCMV感染で1日に上昇してくる分子を調べると、IRF8とZbtb32というセルサイクル制御分子が連れてきた。IRF8の誘導シグナルはIFN系ではなく、IL-12---IL-12R---STAT4を介していることがChIP-seqでも示されている。それだけでなく、MCMVに感染した細胞に発現しているm157に対する受容体Ly49Hを介して、NK細胞のIRF8も上昇するというIL-12Rからのシグナルと二段構えになっているようだ。で、結局IRF8が機能するとZbtb32を介してNK細胞増殖が増強し、抗ウイルス感染機能が上昇する、ということ。こういくクロストークしている受容体を評価するって難しいと思う。
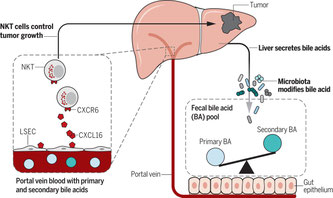
2018年5月28日(担当:横須賀)
Science 25 May 2018: Vol. 360, Issue 6391, eaan5931 DOI: 10.1126/science.aan5931
Gut microbiome–mediated bile acid
metabolism regulates liver cancer via NKT cells
抗生物質を内服していると原発性や転移性の肝臓がんが何故抑えられてるか、というScience
Article。食生活と消化器がんの関係は、細菌の腸内細菌の解析から、そのフローラの変化によるものと考えられているが、肝臓がんはどうか?さまざまながんのマウスモデル(Myc-on、胸腺細胞種EL4皮下注、メラノーマB16脾臓注、B細胞リンフォーマA20静注)の肝臓がんは、混合抗生剤(バンコマイシン+ネオマイシン+チエナム)の内服で軽減する。肝臓の免疫細胞を調べてみるとNKT細胞が特異的に多くなっている(他にもCD8+T細胞やメモリーCD4+T細胞、メモリーCD8+T細胞がそうらしい)。このNKT細胞は肝臓に局在するものとして、CXCR6を発現し、肝臓にいるCXCR6+細胞の70%はNKT細胞である。抗体によるT細胞除去、CD1dKO、CXCR6KOマウスを用いると、どうしてもCTLではなくてNKT細胞が肝臓がんを抑えているという結果。じゃあなぜNKT細胞が多くなっているか、それは、CXCR6ケモカイン受容体のリガンドCXCL16が類洞内皮細胞に発現していて、門脈系からのNKT細胞のリクルートに一躍かっているが、一次胆汁(TCA、beta-MAC、T-beta-MCAなど)が多いとCXCL6の発現が増加する一方、二次胆汁(TDCA、T-omega-MCAなど)によって抑制される。よって、二次胆汁を飲ませたりすると肝臓のNKT細胞が減る訳である。二次胆汁は、腸管内でバクテリアが一次胆汁を酵素処理し精製するモノであって、脂肪酸と結合した後に二次胆汁ごと吸収されて門脈系に入り類洞にたどり着く。そこでCXCL16発現を低下させるわわけである。混合抗生剤の内、バンコマイシインが特に重要で、その特徴としてグラム陽性菌に焦点が絞られ、バンコマイシン内服によってクロストリジウム属が著明に減少することが16SrRNA解析で分かった。特にクロストリジウムが持っている、7α-dehydroxylation
reactionという酵素反応が必須で、それによって二次胆汁が精製される。バンコマイシン投与ークロストリジウム減少ー二次胆汁低下ー類洞CXCL16上昇ーNKT細胞増加ー肝臓がん抑制という流れである。最後に、ヒトの肝細胞がんや胆管がんの患者さんでも、一次胆汁とCXCL16 mRNAの相関のあることが示されている。
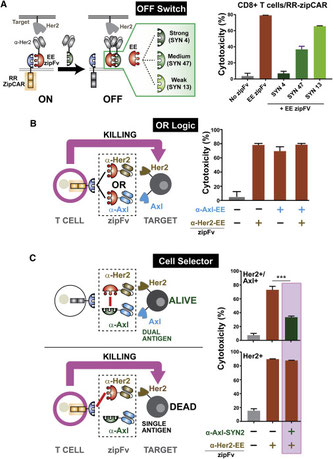
2018年5月21日(担当:矢那瀬)
Cell. 2018 Apr 17. pii:
S0092-8674(18)30362-3. doi: 10.1016/j.cell.2018.03.038.
Universal Chimeric
Antigen Receptors for Multiplexed and Logical Control of T Cell Responses
エンジニアリングのエンジニアリング、CAR-Tの改良のアイデアの論文。細胞外の抗体部分scFvとStalkー細胞膜領域ー細胞内領域(CD3z+CD28+4-1BB-mCherry)とをLeu zipperで繫ぐアイデア。① scFvのアフィニティー、② Zipperのアフィニティー、③ scFvの濃度、④
Zip-CARの発現、の4因子を変えることでCARのチューニングが行える。違う抗体に挿げ替えるられること、正常細胞に発現している分子に対するscFcを使ってZipを中和出来ること、CD3zとCD29+4-1BBとのシグナルを二種類のscFvを用いて分けられること、CD4+T細胞とCD8+T細胞とを別々のscFvで刺激が入れられること、など、アプリケーションが沢山あると。で、①よりも③の方がsensitiveだと。scFvが抗原に結合するのが先か、Zipperが結合するのが先か、さまざまな局面での分子メカニズムは良く分からないが、アイデアとしてまあそういうのもアルよな、と云う感想。
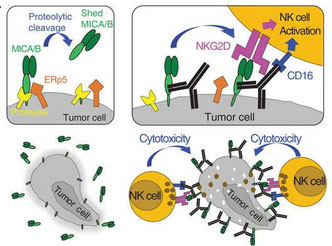
2018年5月16日(担当:秦)
Science. 2018 Mar 30;359(6383):1537-1542. doi:
10.1126/science.aao0505.
Antibody-mediated inhibition of MICA and MICB
shedding promotes NK cell-driven tumor immunity.
活性化型NK受容体NKG2Dのリガンド、MICA/BはプロテアーゼやERp5などによってシェディングされることが知られている。このシェッディングを抗体で阻害したところ、NK細胞による抗腫瘍効果が上昇したというKai W Wucherpfenningの論文。MICA/Bの3つのIgドメインα3を、マウスに免役して3つのクローンを作成、その内の7C6(IgG2a)を使ってマウスのvivoの実験を行っている。抗腫瘍効果は、①主要にリガンドであるMICA/Bの発現がシェディングされずに高く維持されることによるNKG2Dを介したNK細胞活性の亢進、②IgG2aが腫瘍細胞に結合することによる、CD16(FcγRIII)を介したマクロファージの活性化、の2点で、どちらも同じくらい貢献しているようだ。MICA/Bを強発現させた実験系でないと差が出にくいことや、①と②の合わせ技でようやく見栄えのある差がでてくるなど、NK細胞による抗腫瘍効果がどれだけあるのか疑問なところであるが、抗体投与により腫瘍に浸潤している活性化NK細胞が、通常のILC1とは異なることをSingle cell RNA-seqで示したり、NSGヒト化マウスにヒトNK細胞を移入してこの抗体の抗腫瘍効果を見たり、Wucherpfepenningらしい緻密な実験が、がん免疫分野の論文とは一線を画していると思う。
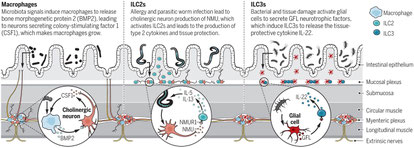
2018年5月1日(担当:若松)
Science. 2018 Mar 2;359(6379):1056-1061. doi:
10.1126/science.aan4829.
β2-adrenergic receptor-mediated negative
regulation of group 2 innate lymphoid cell responses.
粘膜に分布する神経細胞とそれから分泌される神経伝達物質により免疫細胞が刺激であれ、その粘膜またその免疫細胞特有の病原体に対する応答が起こる、というDavid Artisの論文。腸管粘膜にいるILC2とILC3のRNA-seqのデータからILC2にアドレナリン受容体(beta2AR)が高発現していることから始まる。マウスでもひどでも大腸や肺のILC2で高発現していて、絨毛や粘膜下層のアドレナリン作動性神経節にILC2が近接しているようだ。Adrb2-/-ではILC2応答が増加し寄生虫排除が亢進し、b2ARを刺激するClenbuterolを添加するとその逆になる、つまりb2ARシグナルはILC2を抑制しているらし。抗CD4抗体でヘルパーTを除去しても影響はなく、IL-7R-Cre x Adrb2f/fで造血細胞特異的にとばすとILC2が増加することから、どうやらTh2 Tではなく、ILC2が責任細胞らしい。気道粘膜上皮でもNippostrongylus brasiliensis感染+IL-33経鼻投与で同様の結果が得られた。Nippo感染ありなしのRNA-seqの結果からどうやら細胞増殖に関わる遺伝子がb2AR刺激で低下するらしい。結論的には、神経末端から放出されるカテコラミンはILC2の増殖を抑制し、寄生虫感染を助長する(逆にアレルギーは低下するのだろう)、ということ。昨年のNatureの3つの論文(Henrique Veiga-Fernandes、David Artis、Vijay Kuchroo)、神経ペプチドNeuromedin(NMU)がILC2(NMU受容体1を持つ)に働き、ILC2からIL-5とIL13の産生を促し、寄生虫感染に抵抗性を持つ、とは逆の話。神経はILC2を正にも負にも調節しているってことでしょうね。
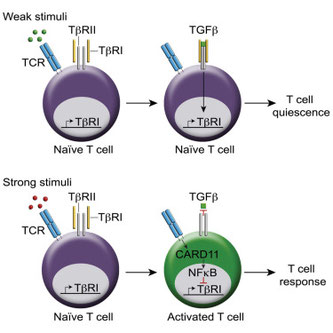
2018年4月23日(担当:町山)
Immunity Volume 48, Issue 4, Pages
745-759.e6 (April 2018) DOI: 10.1016/j.immuni.2018.03.025
T Cell Receptor-Regulated TGF-β Type I
Receptor Expression Determines T Cell Quiescence and Activation
メッセージは、ナイーブT細胞がTCRからではなくTGFbtype1受容体からのシグナルによって積極的にQuiescence静止状態を維持している、ということ。SuboptimalでTCR刺激を加えると特にTGFbRIの発現が低下(何故かmRNAは変わらず、タンパク質レベルで低下)。5C.C7Tgの抗原刺激でも抗原濃度を少なくするとFoxp3発現が上がり、強いとTh1やTh17サイトカイン産生細胞が増える。NFkBやNFATの結合サイトがTGFbRIのプロモーター上にあり、NFkB阻害剤を加えるとTGFbRIが上昇したことから、恐らく強いTCRシグナルがその転写を抑えているのだろう。CAPE阻害剤の投与で、その強いTCRシグナルとはRelA特異的NFkBシグナルがTGFbRIの発現を抑制することが判明。逆にTGFb添加によってSmad2依存的にTGFbRIの発現は上昇する。またここにIL-6を添加すると戻ってしまう。ナイーブT細胞にTGFbRIを強制発現させるとQuiescenceなT細胞増殖と自己免疫は抑えられるし、SLEの患者さんもナイーブT細胞でのTGFbRIの発現が低下しているため、Quiescenceを維持できないことと相関していると。うむむ、ただ、この論文で言及しているナイーブT細胞は一度T細胞を刺激した後のRestingの細胞、本当の意味でのナイーブではないし、エフェクターT細胞にTFGbシグナルをつよくするよう強制したら、そりゃ抑制状態になるだろう。
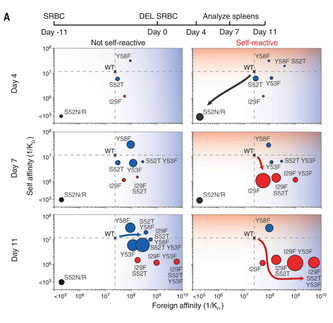
2018年4月16日(担当:横須賀)
Science 13 Apr 2018: Vol. 36 0, Issue 6385, pp. 223-226DOI:
10.1126/science.aao3859
Germinal center antibody mutation trajectories are determined by
rapid self/foreign discrimination
自己反応性を持つB細胞が末梢性寛容の下に存在する意義は何か?という生物学的な問題に対する1つの回答を示したChris Goodnowの論文。カンピロバクタ−とヒトの神経細胞の糖脂質は似ており、0.1%存在すると云われる交叉反応抗体が、感染症を引き金としてギランバレー症候群を発症させる。なぜそんな危なっかしい抗体が必要なのか?普通よりも親和性の低いhen egg
lysozyme(HEL)、HEL3xの膜結合型タンパク質トランスジェニックマウス(Tg)を自己抗原発現マウスとして、そこにHyHEL+ B細胞を移入すると末梢性寛容が起こる。一方HyHELの外来抗原としてHEL3xよりも2倍親和性の低いduck egg lysozyme(DEL)を外来抗原とする。HEL3xTgに骨髄移植されたHyHEL
B細胞はIgD+のアナジーB細胞として末梢に出て来る。このマウスにHEL3xで免疫しても反応性は低いが、DELで免疫すると、HEL3xのnon-Tgと遜色なく反応してしまう。あれ?これでは自己のHEL3x抗原にも反応してしまうのでは?実は1週間の間に胚中心で体細胞超変異が起こり、HEL3xに対する自己反応性は低く、DELの外来抗原には反応する抗体へと変わっている。これは記憶B細胞として血清中の抗体にも残る。しかも、HyHEL+
アナジーB細胞とポリクローナルな正常ナイーブB細胞とをRagKOマウスに移入しDELで免疫すると、HyHEL+
アナジーB細から効率的にDEL反応性B細胞が分化してくる。ギランバレー症候群の例もそうだが、HIVやLCMV、ラッサ熱ウイルスなど、ヒトのタンパク質に疑似したタンパク質分子を持つことでヒトの免疫系から逃れようとしている病原体に対しては、自己反応性が多少あったとしても、末梢性寛容でコントロールし、いざという感染時にクイックに効率的な病原体に対する抗体を産生できるB細胞レパトアを残した方が生物学的に良い、という証拠だとのこと。HELの系でしか証明していないし、そういうアナジーの分画がどの位の数存在し、それがポリクローナルな分画よりも本当に効率的か、は明確ではないが、「なるほど」と思ってしまう。
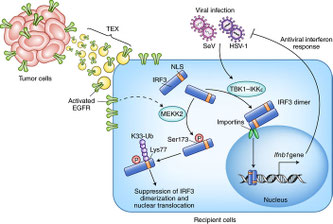
2018年4月9日(担当:矢那瀬)
Nat Immunol. 2018 Mar;19(3):233-245. doi:
10.1038/s41590-017-0043-5.
Tumor-derived exosomes antagonize innate antiviral
immunity.
腫瘍由来のエクソソーム(Tumor-associated exosome :
TEX)が運んできたEGF受容体が、マクロファージの抗ウイルス作用にアンチに働く、という論文。通常、センダイウイルスや単純ヘルペスウイルスなどがマクロファージに感染すると、細胞内DNAセンサーの下流でIRF3の活性化が起こる。TBK1-IKKepsilonを介してIRF3のリン酸化と二量体化が起こると、核内に移行したIRF3によってIFNbが産生され、抗ウイルス応答となる。TEX由来のEGFRの下流で働くキナーゼとして、Kinome
screeningによってMEKK2であることが分った。その結果、通常のIRF3のリン酸化ではないSer173のリン酸化が起こり、二量体形成が阻害され、またIRF3の核移行シグナルシークエンスにあるLys77がK33連鎖のポリユビキチン化を受け、核移行も出来なくなる。MEKK2KO(Map3k2-/-)マウスでin vivoでの生存や、IRF3 S173A Crisper
KIを用いて生理的量での実験など、詳細に検討されている。エクソソームの抽出に使っている腫瘍株LLCはEGFR変異はないようだが、それがEGFR変異によるTKI感受性のあるなしでのマクロファージに対する自然免疫応答抑制の差はみてはどうかと思うし、今後、IRF3 KIマウスを用いて、ウイルス感染に対する新たな治療法開発のin vivo実験を進めている予感がする。N&Vは北大高岡先生。
2018年4月2日(担当:秦)
J Exp Med. 2018 Feb 27. pii:
jem.20171285. doi: 10.1084/jem.20171285.
Peripherally derived T regulatory and
γδ T cells have opposing roles in the pathogenesis of intractable pediatric epilepsy.
癲癇のフォーカスに浸潤している制御性T細胞とgdT細胞の数が、その病状に規定因子になっている、という論文。ラスムッセン脳炎など、小児の癲癇と末梢白血球の脳内浸潤との関係が言われているらしい。治療としてフォーカスを切除する方法がとられているという背景から、4種さまざまな様式の癲癇患者から採取した、フォーカスとその周囲の脳組織に関して、免疫細胞のキャラクターを調べた。するとマイクログリアに差がない一方、マクロファージなどの抗原提示細胞、abT細胞(CD8T細胞の比率が上昇)、gdT細胞の数が多くなり、活性化マーカーも上昇しているようだ。一方、制御性T細胞の数は少ない。カイニン酸腹腔注射による慢性癲癇マウスモデルを用いても、同様の結果が得られ、gdT細胞欠損で症状改善(発作までの時間が短縮され、持続時間も延長する)、制御性T細胞欠損で症状悪化が見られる。癲癇発作重症化のエフェクター分子としては、gdT細胞から産生されるIL-17やGM-CSFなどの炎症性サイトカインらしく、こういったサイトカインにニューロンを浸すと、電気刺激によるスパイクが頻度を増し、結果的にアポプトーシスも誘導される。あくまでサイトカインを産生するのは脳内浸潤した免疫細胞であるから、北大の村上先生が仰るような脳神経活動が発端として起こる炎症のGatewayとは異なるようだ。
2018年3月27日(担当:豊田)
Nature. 2018 Mar 15;555(7696):382-386. doi: 10.1038/nature25974.
Recognition of DHN-melanin by a C-type lectin receptor is required for immunity to Aspergillus
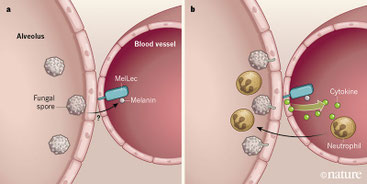
C-type lectinの1つMelLec(Clec1A)がアスペルギルスの産生するメラニンを認識してアスペルギルス感染症に対する免疫に寄与する、という論文。Clec1Aに結合するものとして、アスペルギルスの分生子がとれてきたらしい。Clec1Aは黒色真菌にも結合するが、カンジダや出芽酵母には結合しないし、アスペルギルスも生殖時期のものには結合しない。アルカリ不溶性のものには結合しないため、他のCタイプレクチンとは違って、糖鎖に結合するのではなさそうだ。で、アスペルギルスが合成するメラニン、その中でもジヒドロキシナフタレン環(DHN)を持つメラニンであることが示された。Clec1Aは、骨髄系細胞では発現しておらず、どうやら内皮細胞、特に肺の内皮細胞(CD31+ではあるがEpCAMは陽性陰性関係ない)に発現し、DHNメラニンと結合すると、ケモカインCXCL1を分泌し、好中球の遊走を介してアスペルギルスに抵抗する。ただし、Clec1a KOマウスではBALF中の好中球やCXCL1、GM-CFSは低下するものの(4時間では差があるが、24時間では同等になってしまう)、生存率には関係ない。一方、グルココルチコイドを投与し、さらにアスペルギルスを静注すると、KOでは生存率が下がることから、恐らく免疫不全状態における重症感染時にはClec1aの抗アスペルギルス作用が顕性化するのだろう。in vitroのデータまでは非常に面白いのだが、in vivoのデータの出し方が強引。さらにヒトの例としてアスペルギルス感染に感受性のあるSNPとしてClec1AのG26Aがあるそうだが、骨髄幹細胞移植でドナー側のG26A SNPが重要、あれ?骨髄系細胞には出ていなかったのでは?と矛盾しており、少し無理矢理かなと。恐らくClec1Aがアスペルギルスのメラニンを認識していることは確かに思うが、アスペルギルス感染でのImmunityにどの程度寄与しているか?と考えると僅かであり、重症感染症で真菌血症を起こしているような場合に限定されるようだ。ただ、自然免疫受容体はその種類の多様性故に、合わせ技で効いていれば良く、少しながらでも機能的であれば良いのかも知れない。
2018年3月19日(担当:永井)
J Exp Med. 2018 Mar 5;215(3):815-825. doi: 10.1084/jem.20170901.
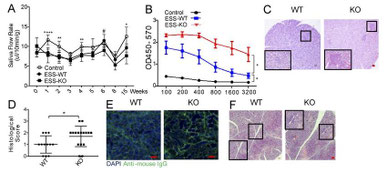
Deficiency in T follicular regulatory cells promotes autoimmunity.
濾胞性御製T細胞(TFR)を欠損すると自己免疫応答が亢進する、というChen Dongの論文。Bcl6f/f x
Foxp3-Creを作成してインフルエンザ感染モデルで実験してみると、TFHのIFNg産生があがり、IgG2にスイッチ、Fluへの抵抗性が上がっている。30週齢で自己免疫疾患が発症し、唾液腺、肺、膵臓への細胞浸潤と腎の抗DNA抗体によるDepositが沈着。ESSのシェーグレン疾患モデル(磨り潰した唾液腺で免疫)でも唾液のフローが低下。ただし、Bcl6f/f x
CD4-CreをESSモデルで試してみると、PD-1+ CXCR5+ TFHとFas+ GL7+のGerminal center B細胞が消失、IL-17細胞が低下してシェーグレン症状も治まると。ううん、予想通りの結果。
2018年3月12日(担当:若松)
Nat Med. 2018 Mar 5. doi: 10.1038/nm.4501.
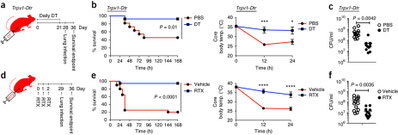
Nociceptor sensory neurons suppress neutrophil and γδ T cell responses in
bacterial lung infections and lethal pneumonia
神経の侵害受容器は有害刺激を感知するが、この手の肺の感覚神経は何をしているんだろうか?侵害受容器などC線維に発現しているTRPV1(Transient receptor potential vanilloid 1)イオンチャネルは、カプサイシン、陽イオン、熱刺激などの反応し、TRPV1発現神経細胞はOVA誘導アレルギー気道過敏に寄与することが分かっていた。Trpv1-DTRマウスでTRPV1発現神経細胞を欠損させると、MRSA感染モデルでのマウス死亡率やショックが改善する。TRPV1の高親和性リガンドRTXを加えTRPV1発現神経細胞を欠損させた実験系でも、肺への細胞浸潤が増え、MRSAコロニー数は減り、炎症性サイトカイン産生が上昇する。浸潤細胞はLy6G+ Gr1+の好中球で、TRPV1発現神経細胞欠損ではより肺での好中球数が増え、動きも良くなり、逆に抗Gr1抗体で除去するとマウスはMRSAで死亡する。さらにTRPV1発現神経細胞欠損では肺のgdT細胞の数が増え、生存曲線もgdT細胞によるらしい。肺の感覚神経支配はほぼ迷走神経であるため、Trpv1-DTRマウスの迷走神経と後根神経節と別々にDTXを局注して潰したところ、迷走神経のTRPV発現神経細胞が責任細胞であることが分かった。侵害受容体を持つ神経が神経ペプチドを放出して免疫細胞を制御すること、その1つCGRP(calcitonin gene–related peptide)がマクロファージからのTNFa産生や皮膚感染でのリンパ節肥大を抑制するという過去の報告をベースに、またMRAS感染によってBALFのCGRPが上昇していたことから、これに注目。CGRPの競合ペプチドでCGRPを阻害すると、TRPV1発現神経細胞を欠損と同じ表現型になるった。つまり、TRPV1↓---CGRP-↓--好中球サーベイ↑+gdT数↑---MRSAのクリアランス↑の軸があると。簡単に言い換えれば、痛いと免疫が下がる、ということだろう。
2018年3月5日(担当:町山)
Immunity. 2018 Feb
20;48(2):299-312.e5.
Interleukin-10 Directly
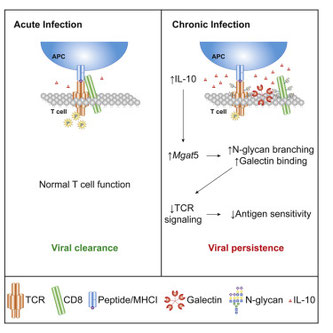
Inhibits CD8+ T Cell Function by Enhancing N-Glycan Branching to Decrease Antigen Sensitivity.
慢性炎症が続くと、CD8+T細胞の感受性が悪くなる。IL-12やIFNgに対する感受性は上がっているし、TCRの発現も変わらないのに、何故だろうか?普通、ここで登場するのはチェックポイント受容体がだが、PD-1もLAG3もCTLA-4もTim3も変わらない。実際は、IL-10を介したダイレクトな
CD8+T細胞に対する抑制だ、という論文。LCMVクローン13の感染実験では、TCRとCD8との距離が遠くなってしまうらしい(FRETで)。どうやらIL-10を介してMgat5というグリコシルトランスフェラーゼが発現上昇し、この酵素によってN-グリカンの分子が増加し、Galectin-3との結合が増え、Membrane
latticeを作り、それが細胞膜の糖修飾を受けている分子の相互作用を阻害するようだ。IL-10だけで培養すればイイらしいから、この条件でTCR Microclusterを解析すると、「粗」に見えるのだろうか?
2018年2月26日(担当:横須賀)
Cell. 2018 Jan
25;172(3):534-548.e19. doi: 10.1016/j.cell.2017.11.037.
Natural Killer Cells
Control Tumor Growth by Sensing a Growth Factor.
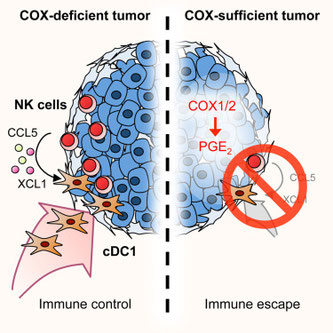
がん免疫応答において、最初にNK細胞が腫瘍浸潤し、次にcDC1が腫瘍微小環境(TME)に呼び寄せられ、腫瘍に対する細胞性免疫応答が亢進する、というSousaの論文。その際、NK細胞はcDC1の遊走に関わるCCL5とXCL1を分泌し、プロスタグランジンE2(PGE2)はこれらケモカインリガンド産生を抑えることで、がん免疫に対峙する。PGE2欠損腫瘍(Ptgs1-/- Ptgs2-/-)ではcDC1(CD8+ CD103+ CD141+ Batf3+ Clex9A+ IRF8+)とNKの腫瘍浸潤細胞数が増加し、お互いの細胞が腫瘍内でクラスターを作る。NK細胞が産生するCCL5もXCL1PGE2欠損で著明に上昇する。BRAF V600E、B16、CT26のどの腫瘍モデルでもNK---CCL5+NCL1---cDC1の関係が成り立ち、皮膚メラノーマ、乳腺浸潤がん、頭頸部扁平上皮がん、肺腺がんのヒト患者でもNK細胞とcDC1の関連性があり生存曲線にも反映している。2015年のCellでSousaらは主張することには、腫瘍の炎症には、COX2により誘導されたPGE2を介した腫瘍増殖性炎症(IL-6、CXCL1、G-CSF)と、PGE2が阻害されたときにおこる腫瘍抑制性炎症(I型IFN、Th1細胞性免疫)との2つがあり、今回そのメカニズムの一部を解き明かした。腫瘍への浸潤はNK細胞ありきで、それがcDC1を介してCTLを誘導するという関係性は、昨年12月のCellでFacundoが示した、インフルエンザ感染において胚中心形成に必要なIL-4は最初のリンパ節にいたNKT細胞が供給する、という話と似ている。やはり自然免疫細胞はさまざまな状況で獲得免疫細胞が活躍する場所作りをするのだろう。
2018年2月5日(担当:秦)
Cell. 2018 Jan 25;172(3):534-548.e19. doi: 10.1016/j.cell.2017.11.037.
Natural Killer Cells
Control Tumor Growth by Sensing a Growth Factor.
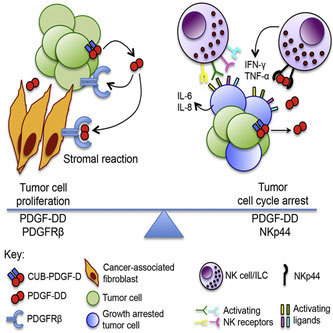
腫瘍から分泌されるサイトカインによってNK細胞が逆に腫瘍の成長を抑制する、というMacro Colonnaの論文。ヒトの活性化NK細胞、ILC1、ILC3、pDCに発現しているNKp44に結合する分子を、NKp44-CD3z/NFAT-GFPの細胞系でSecretome libraryをスクリーニングしたところ、血小板由来成長因子Dホモ二量体(PDGF-DD)が釣れてきた。PDGF-DDは腫瘍からも分泌する分子で、オートクラインにもパラクラインにも、PDGFRbを介して、細胞増殖、上皮間葉転換、血管新生に関わっている。NK細胞は、NKp44を介してPDGF-D or -DDを検知すると、IFN-gやTNF-aを分泌し、Supのみで抗腫瘍活性化を示す。NKp44の下流にはDAP12が会合することが分かっており、PDGF-DDの刺激によっても、Akt-Erk-Foxo3Aが活性化する。グリオブラストーマの例においても、PDGF-DDによるNK細胞の活性化と、腫瘍のGrowth arrestとの相関を認めた。in vivoではどうやら腫瘍から分泌されるPDGF-DDをNK細胞が感知しているようだ。NCR2(PDFG-Dの遺伝子座)Tgマウスでは、抗腫瘍効果や免疫チェックポイントとの相乗効果もあるらしい。両刃の剣のどっちが効いているんだろうか?きっと腫瘍に有利なんだろう。あと実臨床にあるPDGFを標的にした生物製剤って、抗PDFG抗体より抗PDGFR抗体の方がよく効くってこと?
2018年1月22日(担当:永井)
Nat Med. 2018 Jan 8. doi: 10.1038/nm.4466. doi:10.1038/nm.4466
High-dimensional single-cell analysis predicts response to anti-PD-1
immunotherapy.
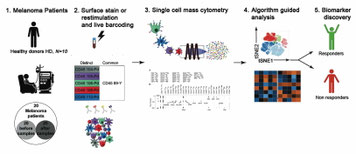
メラノーマに対する抗PD-1抗体療法がどういう患者さんに効果的なのか、その指標をなるマーカーを探索した論文。ただ、メラノーマの患者さんを治療反応群と非反応群とに分け、それぞれ治療前の細胞表面分子がどう違っていたか、CyTOFの結果をクラスター解析しどう相関があるか、という単純でメカニズムもないもの。結果も末血に1.CD14+ CD16- HLA-DRhiのMonocyteが多い群、2.T細胞の少ない群、3.なんでもT細胞活性化マーカーが上がっている群、という訳の分からないの。ただ、もしかしたらNKTが重要なのではないか?と思っている。先週、NKTfhの論文でも、胚中心形成の初期にはTfhではなくてNKTfhが重要、ということだったし、Tumor immunityの初期にもInnate cellの関与が大きいのは理にかなっていると思う。Monocyte---NKT axisが効いているだろうか?
2018年1月15日(担当:若松)
Cell.
2017 Dec 12. pii: S0092-8674(17)31387-9. doi: 10.1016/j.cell.2017.11.036.
Initiation of Antiviral B Cell Immunity Relies on Innate Signals from Spatially Positioned NKT Cells.
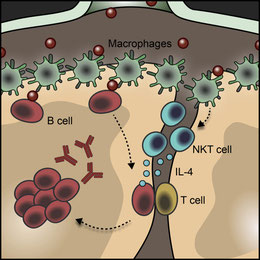
インフルエンザウイルス感染時できるリンパ節(LN)の胚中心(GC)、その形成に必要な初期のIL-4の供給源はNTK細胞からである、というFacundoの論文。CD1dKOマウスにインフルエンザウイルスが感染してもGC形成が悪く、IgG1が出来にくい。どうやら感染後のLNには3日後におこるNKTのwaveと6日後移行におこるTfhのwaveという細胞比のKineticsがあり、NKTはIL-4のPre-Tfh waveを形成する。NKTはCXCR3陽性で、B細胞領域周辺のInterfolicullar areaに集まり、Resident macrophageからのCD1dとIL-18(MyD88を介した下流のシグナルも)を介したヘルプを受け、IL-4を産生する。ジカウイルスのアカゲザル感染でも同じ様な現象がみられていることからも、ウイルス感染時のGC形成過程にTfhが働く以前に、自然免疫細胞がそのプライミングを行っている、と考えると良いようだ。谷口克先生はNKTはやはりTh1 immunityっぽい細胞傷害活性機能が主だ、とおっしゃているけれども、Minor populationではTh2も仕事をしているんだと考えさせられる。FacundoもCarolaとの論文(Identification of Bcl-6-dependent follicular helper NKT cells that provide cognate help for B cell responses. Nat Immunol. 13,35-43, 2012)以降、ずっと考えていたことなんだろう。
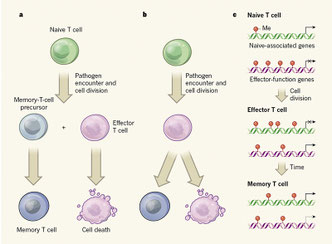
2017年12月25日(担当:町山)
Nature 552, 404–409 (21 December 2017)
doi:10.1038/nature25144
Effector CD8 T cells dedifferentiate into long-lived memory cells.
長期生存メモリーCD8T細胞が、エフェクターT細胞のサブセットから脱分化の過程を介して派生することを明らかにしたAhmedの論文。メモリーT細胞はナイーブT細胞とエフェクターT細胞の両方の特徴を併せ持つ。ナイーブONからエフェクターOFFを経てメモリーOになるような分子にはCCR7やL-selectinなどがあるが、KLRG1+IL7Ra-終末エフェクター(TE)細胞とKLRG1-IL7Ra(CD127)+メモリー前駆(MP)細胞とのナイーブ細胞関連遺伝子およびエフェクター細胞関連遺伝子のDNAメチル化プログラミングの変化を調べると、MPはナイーブなエピジェネティック状態を保持しているというよりも、ナイーブ細胞関連遺伝子がde
novoにDNAメチル化されるプログラムを獲得し、エフェクター細胞関連遺伝子が脱メチル化されることが分かった。ナイーブ細胞関連遺伝子のde
novoメチル化を行っているメチルトランスフェラーゼDnmt3aをエフェクター分化の初期段階で条件的に欠失(Gzmb-Cre)させると、メチル化が低下し、メモリーT細胞の出現が早くなった(=ナイーブ細胞関連遺伝子のメチル化が維持できない)。ゆえに、長期生存メモリーCD8T細胞とは、エフェクター細胞関連遺伝子とナイーブ細胞関連遺伝子とが両方脱メチル化した状態の細胞であり、メモリーT細胞がエフェクター細胞サブセットから生じることになる。ただ、エフェクター細胞でKLRG1を出すか、IL7Raを出すかは、既にそのときに別れていることから
、メモリー細胞が一度エフェクター細胞を通過することは確かだが、ナイーブ細胞の段階で同じものかどうかは分からない。
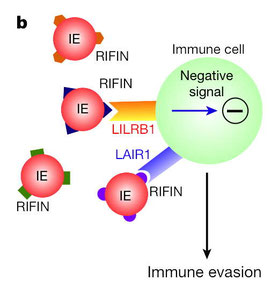
2017年12月18日(担当:横須賀)
Nature 552, 101–105 (07 December 2017)
doi:10.1038/nature24994
Immune evasion of Plasmodium falciparum
by RIFIN via inhibitory receptors
マラリア原虫が抑制性免疫受容体を使ってどのように免疫系から逃避するか、マラリア原虫のRIFINというタンパク質分子がそのリガンドになっているという我が師匠の荒瀬さんの論文。そもそものアイデアは、HSVなど病原体が免疫系から逃避する際、抑制性免疫受容体を刺激して細胞性免疫を抑制する、つまりそのリガンドとなるような分子を発現し、それがペア型受容体の生物学的進化を基板として今のレパートリーを作り上げた、というもの。マラリアも恐らくその機序を使っているだろうと、20種類弱の抑制性免疫受容体の免疫グロブリン(Ig)融合タンパク質分子でマラリア原虫感染赤血球(iRBC)を染色したところ、leucocyte
immunoglobulin-like receptor
B1(LILRB1)という分子が見事に該当した。LILRB1-Igで免疫沈降してマスで解析したらRIFINというiRBCに発現している分子が単離できた。RIFINはPfEMP1やSTEVORと同じように、iRBCに発現しているタンパク質としてアイソフォームが多数知られ(RIFINは150〜200種類も)、マラリアの生活環やアイソフォームで発現がコロコロ変化する。PfEMP1は比較的研究されていて、赤血球や血管内皮の糖鎖や接着分子に結合し、脳マラリアなどの原因となる赤血球の凝集塊を形成する(血管内皮に接着することで脾臓によるiRBCの処理から逃れるのだそうだ)。2015年には、PfEMP1がABO型のA抗原に結合することがわかり、マラリアのエピデミオロジーでなぜマラリア感染地帯でO型が多いのか説明が付いた。で、RIFINのアイソフォームの内、LILRB1に結合するものが見つかり、この結合によってLILRB1のシグナルが伝わること、B細胞のIgM産生とNK細胞の細胞傷害活性が低下することが示された。さらに、RIFINの発現量とマラリアの重症度との間に正の相関があることが、タンザニアのマラリア患者の血液で示され、臨床を反映していることも証明された、、、凄い。マラリア感染地域の住民の抗体を調べると、VとDJとの間にLILRB1が大きくInsertion
mutationで入っている、しかもSomatic hyper mutationをしたIgの方がRIFINとの結合力が増強される、というこの論文のReferにも出て来るLanzavecchiaの論文をさらに読むと、、、MHC、Ig、免疫受容体の「多様性」の魅力=『免疫学の命題』の奥の深さに感銘を受けるのである。
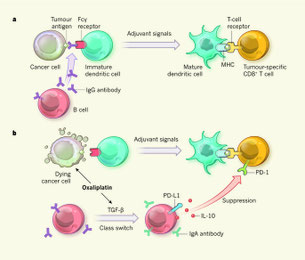
2017年12月4日(担当:矢那瀬)
Nature 551, 340–345 (16 Nov 2017)
A nonimmune function of T cells in
promoting lung tumor progression.
NASHによる肝の慢性炎症と線維化が進むことにより、IgA+ B細胞が肝臓局所に蓄積する。このIgA+ B細胞はPD-L1を発現し、IL-10を産生することで、腫瘍特異的CTLを疲弊化するだけでなく、その肝局在CTLのTCRレパトアを少なくする。獲得免疫系としての抗体があるフェーズでは腫瘍抑制の働を持つ、というMichael Karinの論文。勿論、IgA+
B細胞によって誘導された免疫抑制は抗PD-L1抗体で回復する。またこの免疫抑制機能にはIgA+
B細胞が重要というよりも、血清中のIgAが直接CTLに働くらしい。Iga-/-では活性化したCTLは回復し、腸管にIgAを分泌できないPlgr-/-では血清中のIgA濃度が上昇し、CTLの抑制と腫瘍の増大が起こる。詳細なメカニズムは不明であるが、オリジナルのアイデアは2年前のKarinらの論文"Immunosuppressive plasma cells impede T-cell-dependent immunogenic
chemotherapy." Nature 521, 94–98 (07 May 2015), doi:10.1038/nature14395に由来する(図b)。オキサリプラチンというプラチナ抗がん剤の投与で腫瘍細胞からTGFbが産生され、これがB細胞のIgAへのクラススイッチとPD-L1発現、IL-10産生を誘導する。このとき同時に発表された論文"Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell
immunity." Nature 521, 99–104 (07 May 2015) doi:10.1038/nature14424は、アロ抗体が樹状細胞による腫瘍の取り込みとCTLへの抗原提示を促進するというもの(図a)。B細胞が関連する獲得免疫にも、腫瘍免疫に関して促進と抑制の両方があるらしい。
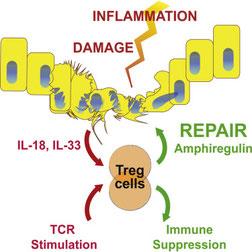
2017年11月27日(担当:秦)
A
nonimmune function of T cells in promoting lung tumor progression.
CD4 T細胞が産生するAmphiregulinが腫瘍増殖を促進させているというRudenskyの論文。インフルエンザ感染後の組織修復が、ResidentialなTregからのIL-18、IL-33刺激(抗原非特異的な応答ではない)によって分泌されるAmphiregulinによる結合織細胞のEGFRを介した増殖による、という自信のCell,
2015を基盤としている。ストーリー的には、Tumor infiltrating Tregが抗原非依存的にAmphiregulinを分泌し、それがTumorのEGRFを介して腫瘍増殖に効いている、ということかと思った。しかし、Foxp3-CreのAmphiregulin conditional
KOで腫瘍の増殖が起こってしまった。しかも、TumorのEGFRをsiRNAでKDしても変わりが無い。結論としてはタイトル通り、TILの中のCD4+ T細胞が出すAmpiregulinが腫瘍微小環境の細胞に働き腫瘍の増大に寄与している、ということ。面白かったが、Rudenskyでもスマートに行かないこともあるのかと思うと、論文の作り方の参考になったし、Tumor
immunityは一筋縄では行かないと再認識した。
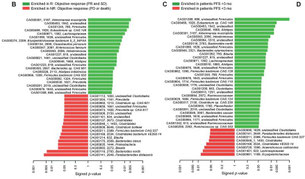
2017年11月20日(担当:豊田)
Science. 2017 Nov 2. pii: eaan3706. doi: 10.1126/science.aan3706.
Gut
microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors.
抗生剤を内服している患者で抗PD-1抗体の効果が弱い、ということからヒントを得て書かれた論文。MelanomaとSarcomaのマウスモデルで抗PD-1抗体による抗腫瘍効果が弱り、またNSCLC、腎細胞癌、尿路上皮癌の抗PD-1抗体治療患者で抗生剤投与群で予後が悪い。NSCLCと腎細胞癌100例の抗PD-1療法反応群と非反応群との、また3ヶ月のProgression-free
survivalのFree群とNonfree群との腸内細菌叢を比較したところ、抗PD-1療法が効く群に優位な腸内細菌として、Verrucomicrobiae網のAkkermansia muciniphilaがピックアップされた。奏効群の患者の便や、この善玉筋を経口投与させると、抗PD-1抗体の効果が増強し、さらにTc1を誘導することが知られていたBacilli網
Enterococcus hirae 13144を添加すると効果は増す。どうやらIL-12依存的なCCR9+ CXCR3+ CD4+ Th1細胞のTumor bedへのリクルートが増強するようだ。タイトルからも上皮癌での話ということが分かるが、Back to backの論文 Gut microbiome modulates
response to anti–PD-1 immunotherapy in melanoma patients. (Science 02 Nov 2017: eaan4236 DOI:
10.1126/science.aan4236)では、反応群の患者の腸内細菌叢ではα多様性(ある1つの環境における種多様性のこと)が高く、Clostridia網のRuminococcaceae科の細菌が優位だと結論している。この違いは癌腫の違いなのか、ともすれば癌抗原と細菌の抗原がクロするのかなどメカニズムは殆ど不明であるが、便治療とチェックポイント療法という二つのトピックが連結する科学的にも医学的にも社会的にも有能なポイントなのだと思う。
2017年11月13日(担当:永井)
Nat Immunol. 2017 Oct 30. doi: 10.1038/ni.3868. [Epub ahead of print]
Oxidative stress controls regulatory T cell apoptosis and suppressor activity and
PD-L1-blockade resistance in tumor.
抗酸化酵素の発現を上昇させるシステムとして、NF-E2-related factor 2-antioxidant response element (Nrf2-ARE) 経路というものがあるらしい。この転写因子Nrf2は、グルタチオンペルオキシダーゼ、ヘムオキシゲナーゼ、カタラーゼなどを発現上昇させる。制御性T細胞 (Treg) は元来Nrf2-ARE経路が弱く、がん微小環境など活性酸素種が多いような条件下では酸化ストレスを受けやすい。一方、これによってアポトーシス関連分子が上昇したTreg (Apoptotic Treg) は免疫抑制効果が返って増強し、それが腫瘍による免疫抑制を助長しているらしい。このApoptotic Tregによる抑制はPD-L1、CTLA-4といったチェックポイント分子や、TGFb、IL-35、IL-10などの抑制性サイトカインを介さず、故に抗PD-L1抗体にも抵抗性である。どうやら、Apoptotic Tregが産生するATPが、Treg上のCD39とCD73との酵素活性を介して代謝され、生成されたAdenosineがエフェクターT細胞を抑制するらしい。ブロッコリーに含まれるSulforaphaneなどの抗酸化作用剤をマウスに投与すると、Apoptotic Tregの作用が減弱し、抗腫瘍効果も増強するため、がん治療には酸化ストレスを除去しましょう、という流れだ。ただ、抗酸化の抗腫瘍効果と、活性酸素種存在下で誘導されるのTregのApoptotic phenotypesとを連結したような論文であって、直接、H2O2など酸化ストレス下で培養したTregの抑制機能が上昇する、というin vitroのデータくらいないと(願わくばin vivoの)どうだろう?と思ってしまう。
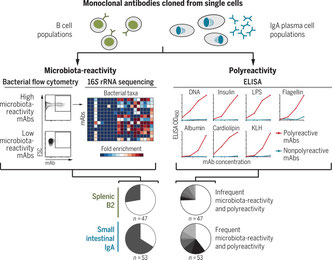
2017年10月30日(担当:横須賀)
Science. 2017 Oct 20;358(6361). doi:
10.1126/science.aan6619
Natural polyreactive IgA antibodies coat the intestinal microbiota.
腸管のIgAの抗原特異性はあまり知られていないようだ。勿論、Pathogenicな病原体が侵入した場合は、それに対する抗原特異性が増すのだろうが、自然抗体として存在するIgAはどうか? Bendelacらは、さまざまなB細胞サブセットのSingle cellから、モノクローナル抗体をIgG1型にしてクローニングし、大スケールでその抗原特異性を調べた。すると脾臓のB-2細胞や辺縁帯B細胞、腹腔B-1細胞に比較して、腸管のIgA産生形質細胞は元々Microbiota-reactiveかつPolyreactiveな抗体が多いそうだ。これは腸管のみでなく、骨髄や唾液腺などIgA産生B細胞に共通した特徴なのである。また、細菌種ではBacteroidetesやFimicutesには反応が低く、ProteobacteriaやActinobacteriaに対しては高いPolyreactivityを示し、細菌のグリカンの種類に志向性があるらしい。またPolyreactivityは、DNA、インスリン、LPS、アルブミンなど様々な物質に対してである。面白いことにこの多様性はSomatic hypermutationには関係せず、よってGerminal centerやT細胞ヘルプを必要としない。これらを必要とするのはIgA産生B細胞の「数としての維持」であって、Microbiota-reactivityやPolyreactivityには関係ない。なぜIgAがこのような特徴を持つのか、メカニズムは全く分からないが、粘膜に共生する細菌叢のバクテリア1個1個がIgA自然抗体によってびっちりコートされている、そんなイメージを具体化させるような論文だった。
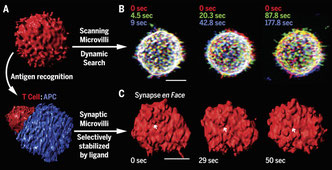
2017年5月22日(担当:町山)
Science. 2017, 356(6338), eaal3118. doi: 10.1126/science.aal3118
Visualizing dynamic microvillar search and stabilization during ligand detection by T cells
T細胞がmicrovilliを用いて抗原提示細胞上の抗原をサーチしている現象を詳細に画像化したMatthew Krummelの論文。ノーベルプライザーのEric Betzigも共著で、Time-resolved lattice light-sheet (LLS) microscopyとQuantum dot–enabled synaptic contact mapping (SCM) microscopyという最新にイメージング機器を使っている。それによると、T細胞がMicrovilliを使って1分程度で抗原提示細胞上のサーチが完了するらしい。一度抗原を見つけてしまうと、全体のサーチの時間が長くなりMicrovilliもスポット的に留まるようになる。ただ、microvilliの単純なサーチは、シグナルや細胞骨格がなくても行われ、先端にT細胞受容体のDensityもまちまちらしい。このMicrovilliは15nm程度の突起らしく、我々が知っているようなNanotuleとはまた別のものらしい。まあ、こうやって抗原提示細胞上にわずかしかないcognateな抗原を探しているのだろう。ただ、まあそうだろう、という現象を詳細にイメージングしただけで、驚きはない。
2017年5月15日(担当:横須賀)
Nature. 2017, 545(7653):243-247. doi: 10.1038/nature22329.
Dominant protection from HLA-linked autoimmunity by antigen-specific regulatory T cells.
Goodpature症候群のHLAとの関連性で、HLA-DR15がsensitive、HLA-DR1がresistantであるメカニズムを、それぞれのHLAアレルが誘導するTヘルパーサブセットの違いから証明した論文。結論的には、HLA-DR1はTregを、HLA-DR15はTh1やTh17を誘導するTendencyがあるらしい。GPのマウス誘導モデルとして、IV型コラーゲンのα3135-145のエピトープを打つモデルでも、このエピトープはマウスとヒトとで共通でもあって、ヒトのアレルの違いでもきちんと証明出来ている。同じα3135-145エピトープを載せる場合でも、HLA-DR1と15とでは、それらの多型性がペプチドをロードするMHC側のポケットに見られ、1アミノ酸ズレて載るらしい。これがTCRレパトアーの違いを生み、TregとTh1/Th17の誘導の違いになっているらしい。こういうMHCの違いで、これだけTCRレパトアに違いがでるのも、珍しいと思うし、データも明確。欲を言えば、TregもしくはTh1/Th17にSkewingするTCRをそれぞれクローニングして、TCR側を決めただけでそれぞれのサブセットに分化出来ちゃう、なんてデータがあればと思う。なぜこのレパトアがTregやTh1/Th17になるのか、そこのところのメカニズムが欠けているから。ところで、荒瀬先生のneo-self仮説では、GPはMisfilding proteinsとは関係ないのだろうか?
2017年5月1日(担当:矢那瀬)
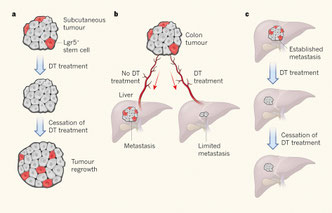
Nature. 2017, 543, 676–680. doi:10.1038/nature21713
A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer.
がんの増殖して進展し転移する過程で、Cancer stem cell(CSC)という概念が重要ならしい。このグループは、マウス直腸がん細胞の中に潜在するCSCが、ロイシンリッチリピートを持つG蛋白質共役受容体Lgr5を発現しており、Lgr5特異的にDTXでこの細胞を除去すると、腫瘍の成長と転移が止まることを示した。Lgr5は本来、大腸のクリプト窩にいる腸管上皮のStemや、毛根、胃壁にいるStemに発現しているらしい。よって、Lgr5+細胞を標的にすれば、がんの増殖や転移を防げるのではないかと。恐らく機能的にも重要な役割をしているだろうが、単なるCSCのマーカーではない点や、他のがん細胞には本当に全く出ていないのか、ということを示してくれないと、CSC以外の細胞は増殖することができない、というおかしなギミックが発生してしまうよねえ。
2016年12月26日(担当:横須賀)
Nat Immunol. 2017 Jan;18(1):86-95. doi:
10.1038/ni.3631.
A cycle of Zap70 kinase activation and
release from the TCR amplifies and disperses antigenic stimuli.
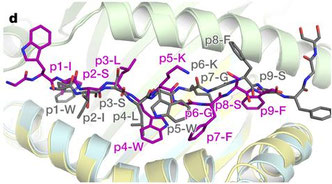
TCRマイクロクラスターに集まったZap70はその後どこにいってしまうのか? という疑問から「活性化したZap70が、TCRマイクロクアスターから連続的に離脱することでTCR活性化シグナルが拡散する」と考えた。Lillemeierは、細胞内 "Serial triggering model" というべき "Catch and release model"
として提唱している。固相化したMHC I-Ek+CD80もしくはOKT3で刺激を行ったT細胞(多くはZap70欠損Jurkat T cell,
P116)の一分子イメージングでは、Zap70の挙動はTCR/CD3とは比較にならない程自由に細胞膜上を動く。タンデムSH2ドメインのみではTCR/CD3と同じ様に固まってしまうため、ヒンジや酵素活性部位が重要で、Mutantの実験からTyr126のリン酸化(これもZap70によるTrans-autophosphorylationによる)とK369のATP結合がZap70のReleaseには必携であると。Zap70
mutantsはTCR/CD3とは異なり、動かないように見えても細胞内との分子の入れ換えはかなり起こっていることがFRAPから分かる。また、このReleaseはT細胞活性化の初期に優位だと。これをCompetition assay、in vitro kinase assay、Biolayer
interferometryなどで詳細に検討している。Zap70のリン酸化のターゲット:細胞膜アダプター分子LATのリン酸化を指標としてアウトプットを検討すると、ReleaseされたZap70によるLATの活性化は条件を絞ればちゃんと起こっていると。ただし、あまりにReldaseが早いとZap70自体のリン酸化が不十分で、そこは兼ね合いのようだ。ただ、pErkやCa2+流入や細胞接着のデータはZap70の挙動との相関性は少ない。本来TCR/CD3は免疫シナプスの中心部へ移動し、その過程でシグナルの活性化と抑制性制御が起こっているため、この論文のようにTCR/CD3を固定した現象が生理的に起こっているとは云えないが、コンセプトの1つとしての提言なのだろう。
2016年12月12日(担当:矢那瀬)
Nature. 2016 Aug 2;537(7620):417-421. doi: 10.1038/nature19330.
Defining CD8+ T cells
that provide the proliferative burst after PD-1 therapy
PD-1抗体療法で抑制解除されるCTLは、CD8+ PD-1+ CXCR5+ Tim3- SCF1+というSubpopulationである、ということを示したRafi Ahmedの論文。LCMV 13株の慢性感染モデルではPD-1hiの疲弊T細胞が出現するが、細胞マーカーの詳細を確認すると、(1) PD-1++ CXCR5- ICOS- Bcl-6loと、(2) PD-1+ CXCR5+
ICOS+ Bcl-6hiの2つのSubpopulationに分けられる。遺伝子発現をみると、後者は2B4やTim3、グランザイム、Id2が低く、Effector
CTLへの終末分化マーカーと云われるものが低く、CD28やICOS、LIGHT、OX40、TLR、TCF1が高いという特色があった。つまり、TFHやメモリーCD8前駆細胞に近く、Th1細胞や終末分化CTLと異なる遺伝子プロファイルを示した。この論文に先行して、既にCXCR5+ Follicular Cytotoxic T cell (TFC)という言葉があるらしい(Nat Immunol,
2016)。CXCR5を発現していることから、この細胞は脾臓やリンパ節などのリンパ組織に移行し、リンパ節に播種しやすいLCMVのクリアランスに寄与しているとRafiは述べる(無理な理論だと感じる)。PD-1+ CXCR5+ CD8+ T細胞を養子移入すると、幹細胞のようにSelf
renewalするし、13株を感染させても増える。恐らく、CXCR5+と-の違いはTCF1の発現の違いに起因するのだろうとのことで、TCF1KO(Tcf7-/-)を用いると、13株感染後のPD-1+ CXCR5+ CD8+ T細胞なくなってしまったそうだ。PD-1+ CXCR5+ CD8+
T細胞の機能解析はなく、現象論を繋げただけで、あまり面白い論文ではないように感じる。よっぽど、Nat Immunol, 2016の方が良いのでは。この論文によると、CXCR5+ CCR7-
TFCはB細胞濾胞に遊走し、B細胞もTFHもTFCも濾胞で出会う。TCFの分化は、TCF1とBcl6とE2Aによって促進され、Blimp1とId2/Id3(E2Aの抑制)は抑制の方向らしい(Conditional KOで)。Blimp1とE2AがCXCR5の発現を規定し(ChIP-seqで)、結論的にBlimp1とE2AがBcl6とTCF1と共にTFCの分化を規定すると。このNat
Immunolの論文が云いたいことには、リンパ濾胞にいるCTL(CXCR5+ CCR7- TFC)は、TFHとCD8 T cell memoryの特徴を両方兼ね備えている細胞であり、EVBなどのウイルス感染で活躍するのだそうだ。一方のRafiは、in vivoの現象論をつなぎ合わせて、PD-1+ CXCR5+
CTLこそが、PD-1抗体療法でブレイクするエフェクター細胞であることを提案したいらしい。両方合わせると良いかも。
2016年11月28日(担当:秦)
Cell. 2016 Sep
8;166(6):1500-1511.e9. doi: 10.1016/j.cell.2016.08.052.
A Distinct Gene Module for
Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells.
Vijay曰く、PD-1+からPD-1+
Tim3+へなるにつれ、腫瘍浸潤リンパ球TILの疲弊度は増すらしい。遺伝子発現を比較すると、疲弊T細胞では亜鉛結合タンパク質Metallothionein(MT)が高値だった。MTは亜鉛などの重金属イオンと結合して、金属元素の濃度調節・解毒・ラジカルの除去を行っているらしい。4つあるアイソフォームの内、ユビキタスに発現している1と2のダブルノックアウトマウスでは、予想通りに抗腫瘍免疫が増大した。WTとMT-/-の健常なCTLと疲弊T細胞とのトランスクリプトーム解析を行うと、MT-/-のCD8T細胞は活性化と機能不全の分子プロファイルがおかしくなっていた。どうしてこんなことになっているのかSingle
cell RNA-seqをしてみると、活性化モジュールと機能不全モジュールとは逆相関にあるようで、そういう機能不全のモジュールもクラスターがあるらしい。で、転写因子に注目してその機能不全のモジュールのトップにランクされた分子から探し出されたのがGATA3とHellios(IKZF2)で、どちらもZink
finger型転写因子であった。GATA3をとばすと当然IFNγやIL-2の発現や抗腫瘍効果は上がり、IL-10の発現は下がる。疲弊状態によるMTと亜鉛がGATA3を介してT細胞機能不全を起こすのだ、という理論。遺伝子発現やプロテオミクス解析だけではなくて、機能的なスコアリングだからこそGATA3がピックアップされてきたのだろう。ただ、数多あるZink
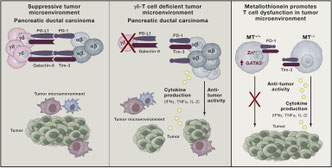
finger型転写因子の中でもなぜ特定の転写因子の機能が影響を受けるのか、こういった場合のT-betとの関係など知りたいことも沢山ある。網羅的解析と既存のGATA3の話をコンバインした感も否めず、煙に巻かれたような気もするし、どなたかこの論文の真意を教えてください。
2016年11月21日(担当:豊田)
Nat Immunol. 2016 Dec;17(12):1415-1423.
doi: 10.1038/ni.3560.
Timing and duration of MHC I positive
selection signals are adjusted in the thymus to prevent lineage errors.
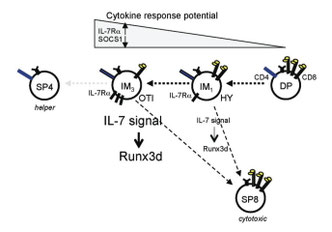
Al Singerのところから継続して仕事を続けていた木村さんの論文。MHCクラスI(MHC-I)拘束性胸腺選択によるCD8陽性T細胞への分化は、2つの正の選択のフェーズに分けられる。フェーズ1はT細胞受容体(TCR)シグナルによる正の選択の開始に始まる過程であり、フェーズ2はサイトカインシグナル、特にIL-7受容体シグナルに依存的なRunx3dの発現誘導によるCD8陽性T細胞への分化の決定の過程である。この2つのフェーズに必要な時間を調べたところ、自己抗原+MHC-I複合体にアフィニティの強いT細胞受容体(具体的にはOT-I TgやCD8.4マウス)を持つ細胞は長いフェーズ1と短いフェーズ2により分化を遂げたのに対し、自己抗原+MHC-I複合体にアフィニティの弱いT細胞受容体(HY Tgなど)をもつ細胞は短いフェーズ1と長いフェーズ2を経て分化を遂げる。つまり、フェーズ1とフェーズ2の進行に必要なシグナルは逆相関しており、その結果、MHCクラスI拘束性の正の選択はほぼ一定の時間で完了する、というもの。フェーズ1とフェーズ2は互いに制御しあうことにより、CD8陽性T細胞への運命決定および分化を効率的かつエラーが生じないよう制御するようだ。Rag2 promoterによって発現誘導されるGFPマウスを用いた、GFPの半減期からのフェーズ1の測定が面白いし、こういう使い方もあると勉強させられる。恐らく、ラインの異なるLck-Cre TgとCre-GFP Tgマウスとの掛け合わせの経験から思い付いたんだろう。それにしても、フェース1のDurationが53時間を境にCD8陽性T細胞とエラーCD4陽性T細胞になるなんて緻密な数字がAl Singerらいしいし、普通じゃ恐くて言えません。
2016年11月14日(担当:永井)
J Exp Med. 2016 Apr 4;213(4):569-83. doi: 10.1084/jem.20151750.
IL-12 drives functional plasticity of human group 2 innate lymphoid
cells.
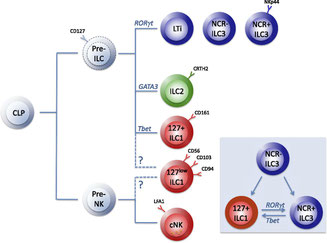
これまでに、ILC3からINFγ産生ILC1への可塑性の報告はあったが(Blood, 124:700-709, 2014)、ILC2に関しては分かっていなかった。ヒト末梢血CRTh2+ ILC2はCD117(c-kit受容体)の発現にもheterogeneityがあったり、細胞表面マーカーも一定しなかったりするのだけれど、T-betの発現が維持されるとIL-13+ IFN-γ+
ILC2となって分化して来るのだそうだ。責任分子のT-betはIL-12—IL-12Rシグナルによって誘導される。IL-12Rβ1欠損を原因する疾患メンデル遺伝型マイコバクテリア易感染症(MSMD)ではILC2への可塑性が失われているし、これまでILC1とILC3しかいないと考えられていたクローン病患者の腸管にもIL-13+ IFN-γ+
ILC2が発見された。Tエフェクター細胞と同じ様に、自然リンパ球も、組織の環境に影響されながら適応した分化をするようだ。制御性ILC(ILCreg)の登場が待ち遠しい、Foxp3だろうかIL-10だろうか。
直ぐ忘れるのでメモっておきます、
ILC1: CD161+ CD117− CRTh2− IFN-γ+ T-BET+
ILC2: CD161+ CRTh2+ IL-13+ GATA-3+
ILC3: CD161+ CD117+ CRTh2− IL-17A+/IL-22+ RORγt+
2016年11月7日(担当:横須賀)
Science. 29 Sep 2016: DOI: 10.1126/science.aaf6284
Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism.
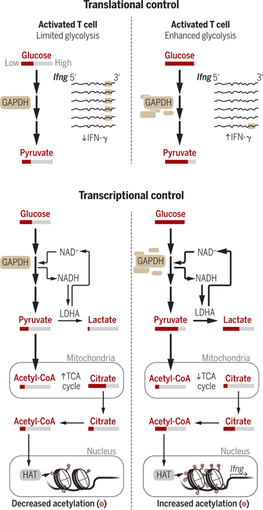
T細胞は、ナイーブ細胞からエフェクター細胞へと分化するに伴い、ミトコンドリアでの酸化的リン酸化から、解糖系によるATP産生へとシフトする。その結果、細胞質で増加したアセチルCoAが、INF-γエンハンサー領域のヒストンアセチル化(H3K9Ac)を促し、INF-γ産生を持続させる。グローバルに云うと、解糖系がEpigenomeを介してTh1の分化を制御しているわけである。きっかけは、1) CD4 T細胞をT刺激すると、乳酸デヒドロゲナーゼ(LDH)の中でも、筋肉型と呼ばれる嫌気性条件下で多く発現するLDHAの増加と、2) 解糖系劣勢の状況では余剰な解糖系酵素グルタールアルデヒド3リン酸デヒドロゲナーゼ(GAPDH)がIFN-γの3’UTに結合し、IFNγの転写を抑制するという2013年のCellの論文。コンディショナルKOマウス:Ldhcf/f x CD4Cre-TgのCD4 T細胞では、解糖系の鈍化と乳酸産生の低下が起こり、解糖系よりも酸化的リン酸化が優位になる。LDHAがないがために解糖系が止まり、その代償として酸化的リン酸化が進まざるを得ず、アセチルCoAもクエン酸として代謝されてしまうようだ。ただ、2013年のCellで示されたようなIFN-γの3’UTRの重要性だけでは説明がつかず、LDHKOマウスでもIFN-γ 3’UTRによるレポーターアッセイでは変わらない。しかし、IFN-γの3’UTRをIRES-eYFPと成長ホルモン3’UTRとで置換したYetiマウスを作製すると、明らかにLDHKOとの掛け合わせでIFNg産生が低下する。IFN-γのエンハンサー領域のChIP-Seq解析をすると、Ldhcf/f x CD4Cre-TgのCD4 T細胞ではH3K9Acが低下しており、面白いことに、酢酸の添加で補償され、クエン酸からアセチルCoAへの合成酵素の阻害で戻ってしまう。先のIFNgの3’UTRがないYetiマウスではIFN-γの産生が維持され、重症の肝障害で生後20日で死んでしまう。しかし、Yeti x Ldhcf/f x CD4Cre-Tgは正常にもどることから、肝障害を誘引するIFNg+ Th1細胞の誘導は、1) IFNgの3’UTRと2) LDHA欠損によるT細胞の解糖系の破綻が起因するH3K9Acの低下による、ことがマウス個体レベルで示された。同じ現象はscurfyマウスとLdhcf/f x CD4Cre-Tgとの交配でも確かめている。学生時代に、解糖系はATPやNADPHなど活性化エネルギー物質のみでなく、核酸・アミノ酸・脂肪酸の材料などにもなる重要な代謝系だ、と生化学で習ったが、ヒストンのアセチル化にも直結しているのは興味深い。なぜIFNgなど特異的な分子のエンハンサー領域でのみヒストンアセチル化が起こるか、次の説明が期待される。
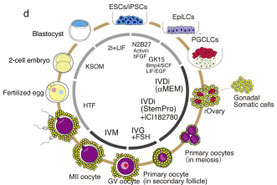
2016年10月31日(担当:古畑)
Nature. 2016 Oct 17. doi: 10.1038/nature20104. [Epub ahead of print]
Reconstitution
in vitro of the entire cycle of the mouse female germ line.
マウス尾の体細胞で作製したiPS細胞から、in vitroの環境のみで成熟した卵子まで分化させることに成功した、という九大林克彦先生の論文。これまではマウスES細胞から卵子をつくることはできても、原始生殖細胞から成熟卵子までの5週間は個体へ移植することによって成功していたが、この過程も培養条件を整えれば完全なin
vitroでできるのだそうだ。データの多くはES細胞を用いたもので、これまでのES研究の歴史の長さを感じるし、iPSのデータは論文にでているのせよ、どこまでESを再現できるのか詳細な比較が知りたい。また、個体発生も、ES/iPS由来の卵子と野生型精子との交配だから、完全にES/iPS由来の精子との受精でも個体発生まで行くのか気になる。30年前に習ったことだが、発生の段階で原子生殖細胞ができて減数分裂と原始/一次/二次/胞状卵胞形成とを経て、人間だったら10年以上かけて成熟卵ができるのが、in
vitroで数週間でできてしまうというのだから、凄い話である。もう分かっているのだろうが、かえって、なぜin vivoではそれだけ時間がかかるのか、なぜヒトの場合は1つずつ成熟卵子ができるのか、Intrinsicな、もしくはNursingしている生殖体細胞による制御(主に抑制)系が知りたくなった。
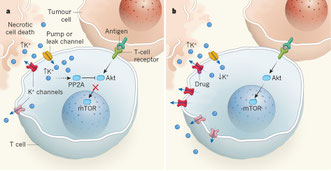
2016年10月3日(担当:矢那瀬)
Nature. 2016 Sep
14;537(7621):539-543. doi: 10.1038/nature19364.
Ionic immune
suppression within the tumour microenvironment limits T cell effector function.
ネクローシスを起こしているような腫瘍組織では組織間液のK+が高く、この高濃度K+がCD8+T細胞(CTL)に作用することで、CTLのIFNγ産生、IL-2産生、細胞傷害活性が阻害される。つまり、TIL(Tumor-infiltrating lymphocyte)が浸潤しているような腫瘍微小環境における免疫抑制が電解質よってなされる=「イオン・チェックポイント」を提唱したいという論文。TILにK+チャネルを導入すると抗腫瘍効果も増強するため、細胞治療にも応用出来るのではないかと結んでいるし、また、K+チャネルの阻害剤を用いても同様の効果が示されている。一方のメカニズムはというと、(pERKとpPLCγ1にK+による変化はないが)Okadaic acidの添加でCTLのK+による機能抑制がリバースされることから、PP2Aを介したpAktの低下によるものだろう、ということだった。ただ、PP2Aのようなユビキタスに効いているようなフォスファターゼを阻害した場合、果たして高濃度K+によるCTL抑制を解除したという直接的な証拠がない。ここはPP2A–Akt–mTORの既存の経路に強引に結びつけた感がある。面白いなと思ったことは、Supplementに出ている、in vitro 培養系にK+を添加するとTregの分化が誘導されること(どこかで既に論文になっているのだろう)。そういえば、TregのT細胞制御のメカニズムの1つとしてConnexin43を介したATPによる話もあるわけだから、意外にK+とかATPとか細胞質にリッチなものは共通してT細胞抑制効果があるのだろうか。インフラマソームとは逆です。
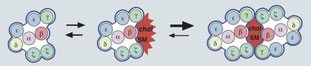
2016年9月26日(担当:秦)
Nat Immunol. 2016 Jul;17(7):844-50.
コレステロール硫酸(CS)の添加がTCRのDimer (Multimer)形成を阻害することを、Mark Davisらしい多様なテクニックで証明した論文。Wolfgang Schamelが提唱していた、コレステロールがTCRb鎖に結合しTCRのMultimer形成を促しているというアイデア(JBC, 2012)と、Resting時にTCR nanoclusterの元となるTCR Dimerが存在するMarkのアイデアとがうまく合致したようだ。CSを添加するとTCRのダイマー形成が阻害されるため、T細胞応答が低下し、CholからCSへの転換酵素Sult2b1の欠損ではChol>CSとなり、T細胞応答が増加する。面白いことに、胸腺DP細胞ではSult2b1の発現が低くCSも少ない、よってDP細胞のTCRシグナルはSencitivityが高くチューニングされているようだ。胸腺に直接CSを注入するとPositive selectionが低下することによるApotosisが起こり、HY-TgとSult2b1KOとを掛け合わせるとNegative selectionが増強する。次の疑問点は、①CSの添加ではなしにTCRのDimer形成を阻害するような実験系でも同様な結果がでるか、②CSを胸腺注入してDP細胞のTCRシグナルをAttenuateした後は末梢T細胞による自己免疫応答がおこるか、かな。Westernの結果を見ると、まだTCRの多くはMonomerのようなので、どの程度のMultimer形成がT細胞応答に呼応してくるかも知りたい所だ。
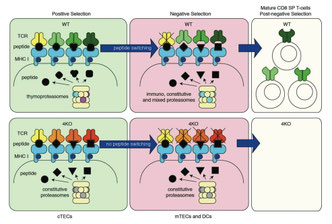
2016年9月12日(担当:豊田)
Nat Immunol. 2016 Aug;17(8):938-45.
Specialized proteasome subunits have an essential role in the thymic selection of CD8+ T cells
免疫プロテオソームに特異的なコンポーネントb1i、b2i、b5iと胸腺プロテアソームのb5tとの4KOを作製すると、特にCD8+ T細胞のNegative selectionで多くがアポトーシスをする、という村田先生・田中先生の論文。そのロジックは、Positive selectionで使われた自己抗原ペプチドが、Negative selectionでも全く同じ自己抗原ペプチドのプールとして使われてしまうため、というもの。新田先生・高浜先生がCD8-restricted TCR-Tgをいくつも掛け合わせてようやくみえてきた胸腺選択の異常が、4KOではNon-Tgで見えています。ということは、5btはClass Iにのる自己抗原ペプチドのバラエティーを増やす為のモノ、という意味合いが強く、b1i、b2i、b5iが補完できているのだとしたら、Stoichiometoricにも各コンポーネントがmixedされたプロテアソームは想像以上に多いのだろう。①ロードされている自己抗原ペプチドが実際違ってくるのかMass spectrometricな解析、②4KOとWTとでのTCRレパトアの相違、③CD4+ T細胞では全くintactな理由、を知りたい。