
Cancer-immunity cycle
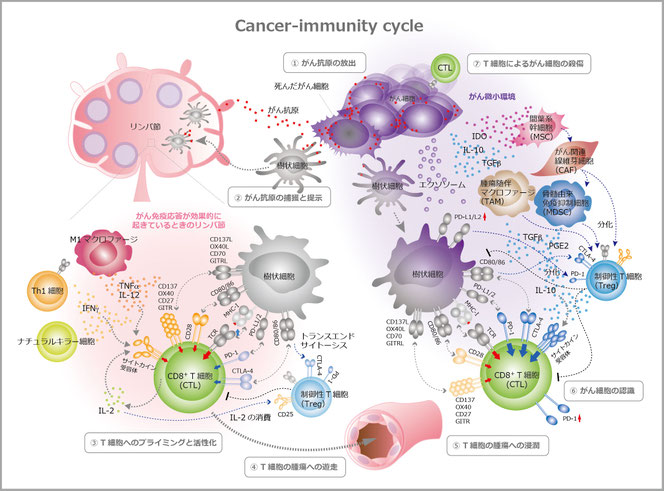
がん免疫が機能しているということは、「がん-免疫サイクル」というがんに対する免疫応答の増幅回路がまわり続けることであり、その結果、T細胞応答は増強し、標的となるがんへの多様性も増大する。このサイクルは7ステップに分けられ、①がん抗原の放出、②樹状細胞によるがん抗原の捕獲と提示、③樹状細胞によるT細胞のプライミングと活性化(図左)、④T細胞の腫瘍への遊走、⑤T細胞の腫瘍への浸潤、⑥T細胞によるがん細胞の認識、⑦T細胞によるがん細胞の殺傷、にて一巡する。このサイクルにはネガティブフィードバック機構も備わっており、過度に抑制されるとサイクルは停止し、がん免疫応答の増幅も止まる(図左)。特に、免疫チェックポイント分子CTLA-4(cytotoxic
T lymphocyte 4)とPD-1(programmed-cell death 1)は、このサイクルのオン・オフを行う免疫スイッチ “immunostat(immune + rheostat)”として機能する。また、免疫抑制因子や免疫抑制細胞が優位になりがん微小環境が構築されても、このサイクルは止まり免疫応答も低下する(図右)。
図左
T細胞によって傷害される、または増殖による壊死や治療によって死んだがん細胞からがん抗原が放出される(①)。がん抗原を捕獲または死んだがん細胞を貪食した樹状細胞がリンパ節に遊走、もしくは、がん抗原がリンパ流によってリンパ節へ運ばれ、そこで樹状細胞により捕獲される(②)。樹状細胞はがん抗原を消化し抗原ペプチドとしてMHCクラスI分子上に載せ、リンパ節内のCD8陽性T細胞に提示する(③)。T細胞はT細胞受容体(T
cell receptor : TCR)を介してがん抗原を認識するが、その際に副刺激受容体である免疫グロブリンスーパーファミリー分子CD28やTNFスーパーファミリー受容体CD137、OX40、CD27、GITR(glucocorticoid-induced TNF receptor family-regulated
gene)が樹状細胞上のリガンドCD80/CD86、CD137L、OX40L、CD70、GITRLそれぞれと結合すると、T細胞に第二の活性化シグナルが伝えられる。M1マクロファージが産生する炎症性サイトカインTNFβ(tumor necrosis factor β)やIL-12、Th1(T helper
1)細胞やナチュラルキラー細胞などが産生するTh1サイトカインIFNγ(interferon
γ)、またCD8陽性T細胞がオートクラインとして分泌するIL-2の作用でさらにT細胞の活性化は亢進する。一方、TCR刺激後半日程度から発現し始める抑制性の副刺激受容体いわゆるチェックポイント分子CTLA-4とPD-1は、抗原提示細胞上のCD80/86やPD-L1/PD-L2とそれぞれ結合し、T細胞を抑制、過度な免疫応答や自己反応性が出ないよう制御している。
図右
がん細胞に上皮間葉転換が起こると、間葉系幹細胞(mesenchyme stem cell : MSC)やがん間質線維芽細胞(cancer associated fibroblast : CAF)介して、骨髄由来抑制細胞(myeloid-derived suppressor cells : MDSC)や腫瘍随伴マクロファージ(tumor-associated macrophage :
TAM)、さらには制御性T細胞の分化が誘導される。このようながん微小環境下ではTNFの他にも抑制性サイトカインIL-10やTGFβ、IDO(indoleamine 2,3-dioxygenase)、PGE2(prostaglandin
E2)などの免疫抑制因子も多く、CD8陽性T細胞上のPD-1や、樹状細胞上のPD-L1/PD-L2の発現が上昇する。このチェックポイント分子を介した抑制シグナルの上昇、また制御性T細胞から分泌される抑制性サイトカイン、CD8陽性T細胞に対する制御性T細胞の直接的な抑制、制御性T細胞による樹状細胞上のCD80/CD86のトランスエンドサイトーシスなどを介して、T細胞の活性化はますます減弱する。
抗PD-1抗体の登場で、がん免疫療法はまさに今ターニングポイントを迎えている。これまでもメラノーマには比較的効果的ながん免疫療法もあったが、免疫原性の高くはない他のがん種への奏効率は高くなかった。しかも、この免疫チェックポイント療法は比較的副作用も少ない1)。
「がん」とは、たくさんの遺伝子異常が蓄積し、正常な細胞制御機能を失った細胞である。「がん抗原」は、そのたくさんの遺伝子異常、特に腫瘍化には関連のないパッセンジャー変異にみられる「ネオ抗原(neoantigen)」や、胎児分化過程でのみ発現する「分化抗原(differentiation antigen)」、がんと精巣に特異的に特に発現する「がん精巣抗原(cancer testis
antigen)」などであり、主要組織適合抗原複合体(major histocompatibility complex : MHC)上に載っている。CD8陽性細胞傷害性T細胞(cytotoxic T lymphocyte :
CTL)がそれを認識しがんの抑制に働いていることは明らかであったが、治療への応用までには至らなかった。その理由としてT細胞からの攻撃を受け続けることで免疫に抵抗性を持つがんへと進化する、いわゆる「免疫編集(immune editing)」があり、T細胞でいう「チェックポイント」があり、腫瘍のメラノーマは「免疫スイッチ(immune rheostat :
immunostat)」がある2)。
がん-免疫サイクルの7つのステップ
免疫監視で一日に3000個自然発生するがん細胞を駆逐する際、またはCTLが腫瘍塊に攻撃を仕掛ける際、がん抗原と免疫細胞応答との相乗サイクル、いわゆる「がん-免疫サイクル(cancer-immuno
cycle)」が知られている3)。がん-免疫サイクルは、①がん細胞からのがん抗原の放出、②樹状細胞によるがん抗原の捕獲と提示、③樹状細胞によるT細胞のプライミングと活性化、④T細胞の腫瘍への遊走、⑤T細胞の腫瘍への浸潤、⑥T細胞によるがん細胞の認識、⑦T細胞によるがん細胞の殺傷の7つのステップで構成される。さらに末梢で誘導される免疫寛容を回避するためには「危険シグナル(Dangerous
signal)」が必要であり、この免疫原性を増強させるシグナルとは、炎症性サイトカインであり、死んだがん細胞からの放出物質であり、腸管細菌叢からの免疫賦活化分子である。
がん抗原は樹状細胞などの抗原提示細胞によって補食され、MHCクラスI分子とクラスII分子上に提示される。ネオ抗原など自己の遺伝子におきた変異は外来抗原として、また胎児がん抗原のように中枢性寛容を逃れた免疫原性のある自己抗原としてT細胞に認識される。このナイーブT細胞のプライミングフェーズにおける、がん抗原特異的T細胞と制御性T細胞の活性化や数のバランスが、最終的にがん免疫応答が惹起するかの鍵となる。エフェクターT細胞(CTL)へと分化すると、CTLは血管内腔へと遊走し、たどり着いた腫瘍部の血管内皮細胞を通過して腫瘍床へと浸潤する。TCRががん抗原+クラスIと結合すると、標的となるがん細胞を認識し殺傷、さらに殺傷されたがん細胞からがん抗原が放出され新たなCTLを誘導、がん-免疫サイクルの活性化のスパイラルによってがん免疫応答が増幅される。このサイクルが回り続ければがんは抑制できるはずであるから、恐らく、がん患者ではこのサイクルのどこかで障害が起きていると考えられる。一方、免疫チェックポイント療法は、このサイクルが正常に回転するよう促す。
がん免疫応答を亢進・抑制する背景因子
がん免疫応答が機能的に起きている環境では、インターフェロンγ(interferon γ)やIL-12、腫瘍壊死因子α(tumor necrosis factor α :
TNFα)が優位な、いわゆるTh1の土壌になっている。そこでは、異物排除に傾倒するM1マクロファージや、細胞性免疫を亢進させるTh1型CD4陽性T細胞、ナチュラルキラー(natural killer :
NK)細胞が機能しており、樹状細胞からもがん抗原の提示と共にCD28など活性化副刺激シグナルが伝えられる。また御性T細胞に比し、CTLが機能的にも数的にも優位である。しかし、一度がん-免疫サイクルが破綻すると、がん細胞から放出されるエクソソームや周囲の結合織からのIL-10、トランスフォーミング成長因子(transforming growth factor β :
TGFβ)、インドールアミン酸添加酵素(indoleamine 2,3-dioxygenase :
IDO)など免疫抑制因子が優位となり、さらにそれらの因子によって間葉系幹細胞、がん関連線維芽細胞、腫瘍随伴マクロファージ、骨髄由来免疫抑制細胞などが分化誘導され、ますます免疫抑制状態となり、がん微小環境を構築することとなる4)。特に抑制性サイトカインによって樹状細胞をはじめ結合識細胞のPD-1およびPD-1リガンドの発現上昇が誘導されると、CD4陽性T細胞からは制御性T細胞が分化され、CTLはPD-1を介した不応答状態、つまり消耗(疲弊)T細胞となる。抗PD-1抗体の投与は、こCD8陽性T細胞が樹状細胞からがん抗原を提示されプライミングされるフェーズで、また最終的にCTLががん細胞上のがん抗原を認識し殺傷するエフェクターフェースで、正常ながん-免疫サイクルへと戻すことを意味している。
がん抗原の提示とT細胞の賦活化
免疫チェックポイント療法が登場する以前は有効的ながん免疫療法が少なかったが、その理由の1つとして、がんワクチンを中心に研究が進んでいたことが挙げられる。感染症撲滅の時代からワクチンは有益な治療法としての確固たる地位を築いてきたが、がんワクチン開発においては二つ課題があった。1つは、CTL応答を出すためにがん抗原をどのように投与すれば最適か、つまり何を抗原にすればよいか、デリバリーの方法はどうすべきか、アジュバントは何にするのか、などハード面での知識が不十分であった点である。2つ目は、がん微小環境自体が“immunostat”として働いている以上、通常の感染症とは異なり、免疫賦活化剤を併用し疲弊や消耗などの環境を改善しない限り、いくらがん抗原のみを投与しても効果が生まれにくいとういことである。ゆえにチェックポイント療法をアジュバントとしてがんワクチンを投与することで、これまで以上の抗腫瘍効果が期待でき、実際、がん精巣抗原の1つMEGA-A3での併用療法が試みられている5)。
しかし、がんワクチンに関しては、まだどの個人にも効果のあるがん抗原の候補が絞れていない。もしドライバー変異となるタンパク質分子ががん抗原になり得るとしたら、それが一番ユニバーサルといえるが、主要なゲノム上の突然変異や、転位、がん精巣抗原などを網羅的に探ってもエピトープを見つけるのは困難であろう。がんの変異は人と人、さらには細胞と細胞で異なるし、見つけたがん抗原が多様なアレルを持つMHCに載るとは限らない。バイオインフォーマティックスやシミュレーションでの探索、MHCから溶出したペプチドのマス解析、という方法もある。また、多価ワクチン合成におけるクラスIIエピトープの必要性も明確な答えがなく、変異を持つ体細胞遺伝子をワクチンに用いたときの自己免疫誘導の問題も解決していない。また投与形態もエマルジョンを用いたり、樹状細胞に直接振りかけたり、養子樹状細胞移植を行ったり、基本的な薬物動態の調査が必要である。
一方、腫瘍が体内に存在するということは,それ自身が内在性の抗原となり、変異やデリバリーの問題なしに必然とこの腫瘍に反応するT細胞が誘導されることを意味する。免疫療法に標準的ながん殺傷療法を重ればさらに相乗効果が期待できる。最近ではがんワクチンをバイパスすべく、キメラ抗原受容体(chimeric antigen receptor ;
CAR)-T細胞療法やTCR-T細胞療法などの遺伝子操作T細胞を患者に戻す試みが行われ、特にB細胞リンパ腫に対するCD19 CAR-T療法が奏効している6)。
以上のように、がん抗原の提示とがん特異的T細胞の活性化は、免疫監視における生理的な反応においても、また治療としてのがん免疫応答の誘導においても要となるステップであり、がん-免疫サイクルを次の段階へと進めるためのさまざまな工夫がなされていること理解できるであろう。
(Trends in CANCER IMMUNOLOGY 2018.7 (1)1より一部改変掲載)